FDA's move toward a single-trial default does not lower the evidentiary bar. It changes where the burden sits. With one fewer trial to absorb uncertainty, reviewers look harder at the evidence that remains — and endpoint selection is where that scrutiny concentrates most.
For surrogate endpoints, the question is direct: why should FDA believe this measure predicts how patients feel, function, or survive? Under accelerated approval, FDA may rely on a surrogate that is reasonably likely to predict clinical benefit. But that likelihood cannot rest on statistical association alone. A defensible surrogate argument requires biological plausibility, empirical evidence, and confidence that the endpoint remains interpretable outside the controlled conditions of a trial. That last requirement is where natural history data becomes essential.
Why trial data alone is often insufficient
Clinical trials are selective by design. Patients are enrolled under narrow criteria, assessed on protocolized schedules, and monitored in ways that bear little resemblance to routine care. A surrogate validated only within that population carries real uncertainty about whether it will perform the same way across the broader group who will eventually receive the drug.
Natural history studies answer a different question: does the endpoint behave coherently across broader patients, irregular assessment patterns, comorbidities, treatment changes, and real-world care pathways? That is the evidence that makes a surrogate argument more than theoretical.
A regulatory-grade natural history study is not just a descriptive registry. Each design requirement connects directly to what a surrogate argument needs to hold up under review:
- Pre-specified analysis plan: Registered before endpoints are derived, so reviewers cannot argue that endpoint choices were driven by the data
- Individual patient-level longitudinal records: Aggregate summaries cannot show how a surrogate tracks disease over time in individual patients
- Complete records across multiple sites of care: A surrogate validated in a single academic center tells you less than a surrogate that holds across community and specialist settings
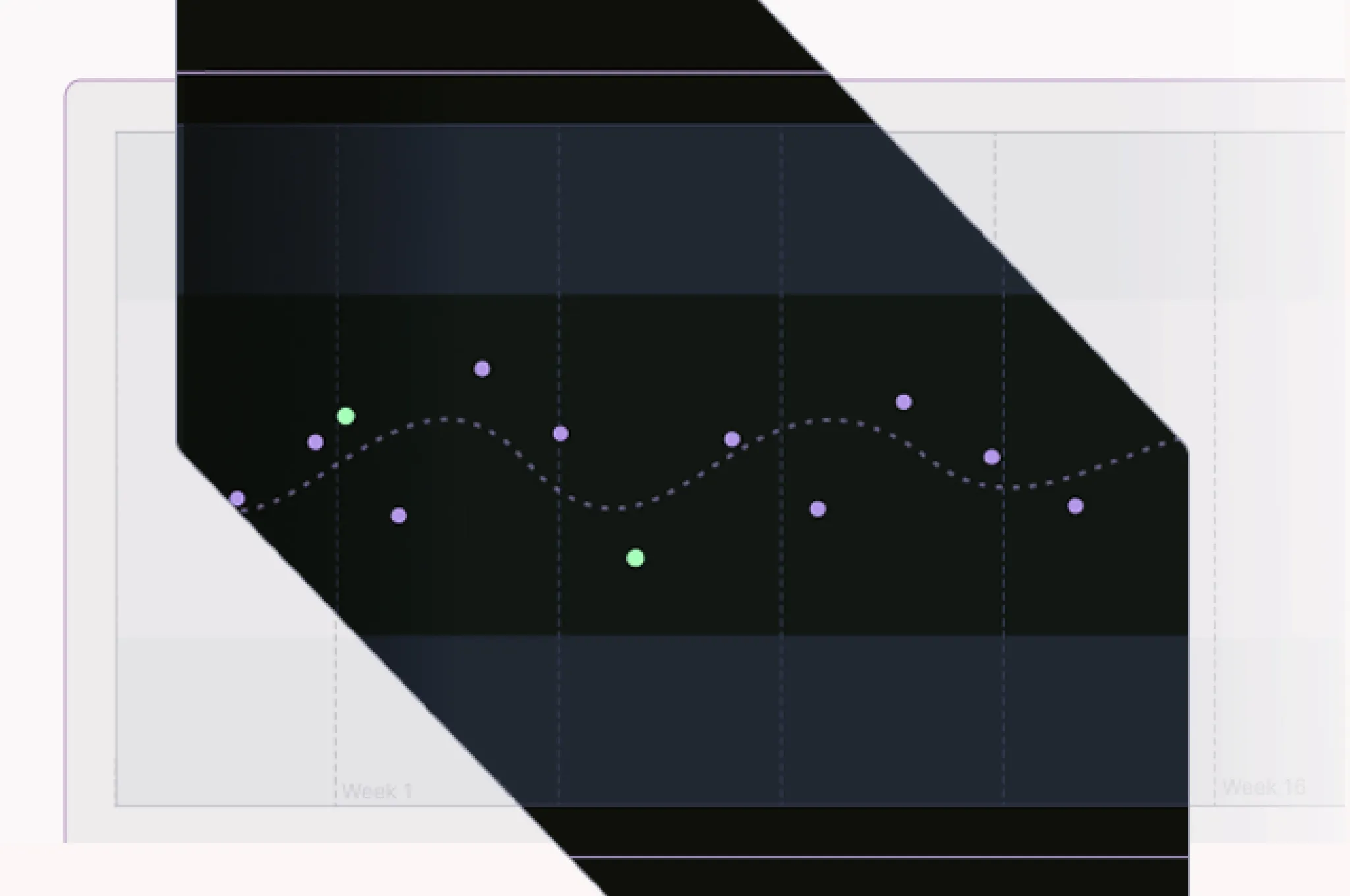
- Follow-up long enough to capture disease trajectory: Behavioral consistency of a surrogate requires showing it moves with the disease over time, not just at a single point
- Retention that avoids attrition bias in trajectory estimates: Patients who drop out are rarely representative; systematic loss distorts the picture of how disease progresses
- Population breadth beyond the trial-eligible subset: Real-world behavioral consistency means the surrogate holds across severity levels and patient types the trial excluded
Natural history studies in practice
Natural history studies solve different problems in different disease contexts. Below, two PicnicResearch studies illustrate the range.
Where no validated surrogate exists: LC-FAOD
Long-chain fatty acid oxidation disorders (LC-FAOD) present a foundational endpoint problem: the disease simultaneously affects cardiac function, energy metabolism, and exercise tolerance, and no validated composite surrogate exists to capture it. The challenge is not refining an existing measure, it is demonstrating that a composite of clinically meaningful events can be constructed and tracked consistently from routine records without a protocol imposing uniformity on how or when data are collected.
The Odyssey study addressed this directly by integrating major clinical events (rhabdomyolysis, hypoglycaemia, cardiomyopathy), lab trajectories, treatment sequences, and linked patient-reported outcomes (PROs). Patients experienced fewer major clinical events and annualized inpatient days during triheptanoin-treated periods than during medium-chain triglyceride oil-treated periods, demonstrating that real-world records can support comparative effectiveness conclusions even where no validated endpoint existed at the outset.
Where a candidate measure is insufficient: hemophilia
In hemophilia, the problem is structurally different. A candidate outcome measure exists — bleed frequency — but it misses severity, treatment burden, pain, functional limitation, and quality-of-life impact. Medical records capture clinical events but not the patient experience of living with the disease; PROs capture that experience but lack the clinical context to make it interpretable to regulators. Neither source alone is sufficient.
PicnicResearch's longitudinal hemophilia registry addressed this by integrating medical records, treatment history, bleeding events, and patient-reported outcomes into a unified dataset. The resulting study demonstrated how multidimensional disease burden can be characterised more completely when clinical and patient-reported data are captured together over time, producing an endpoint picture that neither source could generate independently.
Natural history studies are becoming load-bearing evidence
Natural history studies have often been treated as background context. For single-trial submissions built around surrogate endpoints, they become load-bearing evidence.
The sponsors best positioned for this environment will be the ones who build longitudinal, pre-specified, fit-for-purpose natural history datasets before the pivotal readout — not after FDA asks for proof.
PicnicResearch's direct-to-patient platform retrieves complete medical records from every site of care, structures them into regulatory-grade data regardless of source or format, and maintains 98% annual patient retention — the operational foundation that makes natural history studies feasible at the timescales surrogate validation actually requires.
For more information on how the one-trial default reshapes evidence strategy across the full development lifecycle — from natural history through post-market follow-up — read The Real-World Implications of the FDA's One-Trial Default. For more information on how PicnicResearch can support your natural history study needs, contact us.