The February 2026 New England Journal of Medicine piece from FDA Commissioner Marty Makary and CBER Director Vinay Prasad officially retired the two-trial default for drug approvals, restructuring the evidentiary requirements sponsors must meet before approval.
For oncology and rare disease developers, single-trial approvals backed by confirmatory evidence have been standard practice for years because high disease burden and constrained patient populations have made replicating a pivotal trial impractical, and often unnecessary. The new guidance formally extends that logic to common conditions where the two-trial expectation has historically been enforced most consistently. The goal of the shift is to speed time to evidence and reduce cost overall, a benefit for sponsors, patients and payers alike.
This is a meaningful procedural shift. But it comes with an important qualifier: the FDA retains full discretion to require more research when the evidence warrants it, and single-trial submissions will face closer scrutiny on endpoint selection, control arm quality, effect size, and biological plausibility than two-trial packages typically did. The bar has not been lowered. It’s been moved.
The consequential question is what happens after approval. Reducing the pre-market evidence requirement shifts confirmatory burden to the real world, and the infrastructure to carry that burden is not yet fully built. This is where we think the opportunity lies, and it runs across the entire development lifecycle.
Building the foundation before the trial starts
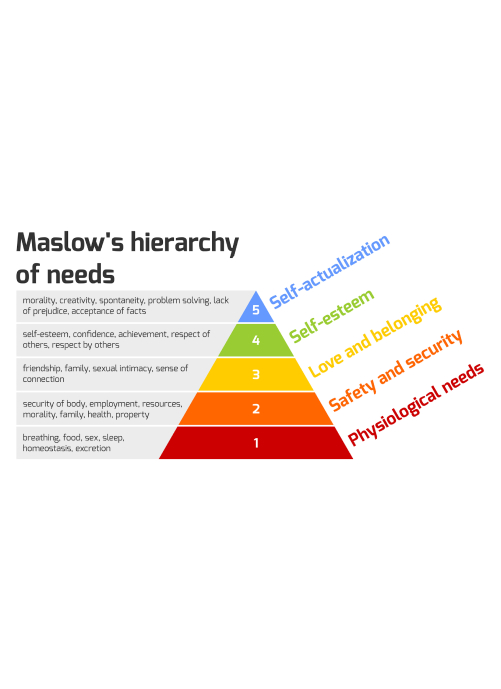
Single-trial submissions live or die on endpoint credibility. When a sponsor argues to the FDA that a surrogate endpoint meaningfully tracks clinical benefit, that argument requires real-world grounding: evidence that the biomarker or intermediate measure actually predicts outcomes in patients who look like the ones who will eventually receive the drug. Natural history data built from longitudinal patient records serves this function, capturing how patients actually progress under standard of care and how candidate endpoints behave outside of controlled conditions.
PicnicHealth’s direct-to-patient evidence platform builds living natural history data assets from consented patients' complete medical records aggregated across every site of care, not just a single institution or claims data feed. The result is a longitudinal, clinically deep picture of real-world disease progression — treatment sequences, biomarker trajectories, comorbidity patterns — that gives sponsors the pre-trial grounding a single-study regulatory strategy demands.
Enriching the enrolled population's story
Patients don't arrive at a trial without history. Comprehensive pre-enrollment records give sponsors the context to understand their enrolled population, account for baseline heterogeneity, and build the biological narrative the FDA increasingly expects in single-study submissions.
Screen failures matter here too. In diseases where eligibility criteria are stringent and biomarker-driven (Alzheimer's disease is one example, where screening failure rates can exceed 50%), the patients who don't qualify for the trial represent a fully characterized cohort that typically disappears from the evidence base entirely. Retaining and following screen failures longitudinally creates both a potential comparator population for external control arm construction and a data source that can sharpen eligibility criteria for future programs. Because PicnicHealth maintains a direct, ongoing relationship with patients and continuously retrieves their records from all providers, screen failures and trial completers alike can remain part of a living evidence asset rather than falling out of follow-up the moment the trial interaction ends.
Sustaining credibility after approval
The post-market confirmatory burden is where the new default policy will ultimately be tested, and that is the largest unknown. Demonstrating that trial results hold in broader, less selected populations requires longitudinal follow-up over timescales that no pivotal trial budget can accommodate, capturing safety signals that spontaneous reporting systems miss, and building the evidence base that supports label expansions as real-world use patterns reveal new questions.
PicnicHealth’s 98% patient retention rate is what makes multi-year post-market follow-up actually feasible. And because our AI-powered platform structures complete medical records into regulatory-grade data across any EHR source or format, sponsors can track the outcomes that matter without being limited to coded fields in claims or registry databases.
The one-trial era doesn't reduce the evidentiary work, it redistributes it. The sponsors who navigate the shift most effectively will be the ones who treat real-world evidence not as a post-hoc supplement, but as a continuous thread running from natural history through approval and into long-term follow-up.
The evidentiary bar hasn't been lowered — it's been moved. PicnicHealth's direct-to-patient platform supports sponsors at every stage of that shift, from natural history to post-market follow-up. Contact us to learn more.